When setting your sort gates in Diva, there are some limitations that users need to be aware of that are not obvious, especially when it comes to gating on populations close to or below the axis.
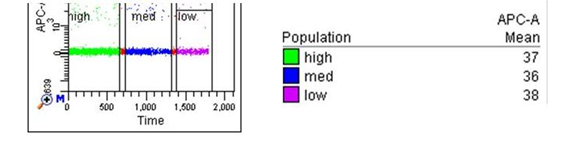
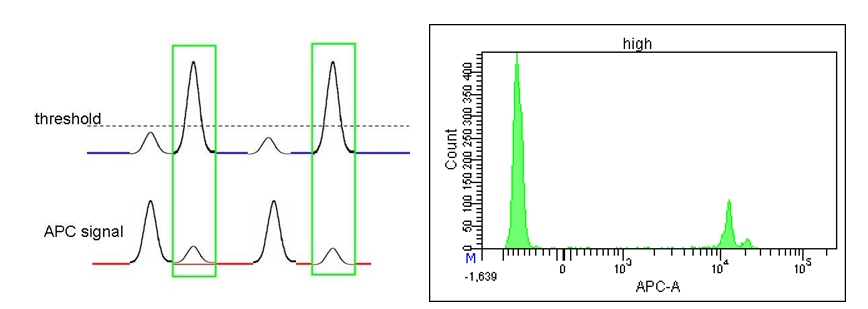
I first encountered this issue when samples were either overcompensated, or (as discussed in the previous post) when small fluorescent particles pushed the baseline restore artificially negative, so that the population of interest was at or below 0. There is a minimum “upper limit” for your sorting gate, and if you set the region boundary below this, no events will be sorted (your sort rate and collected events will be 0, even though you see events in the gate on the plots). This is true for ANY gate in the hierarchy, so if your “sort gate” is a descendant of a gate that is set too low, you will not register any sorted events.

Setting the gate any lower than shown for APC will result in no events being sorted.
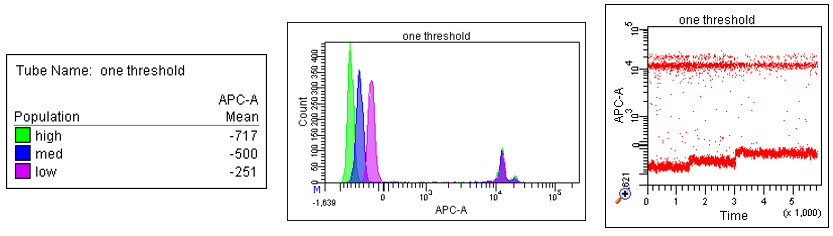
The next limitation to highlight is that Diva cannot distinguish a LOWER boundary that is less than this cutoff—if you are sorting below this minimum limit, then ANYTHING below that is considered “in the gate” regardless of what lower boundary you set. In this example, region P5 is set around only the orange population, but in the post sort analysis on the right, you can see the green population was ALSO sorted.

Lastly, if you were going to try to sort just the green population, you could set at least one point of the gate above that “minimum threshold” value to get around that “minimum threshold” and get events to sort, and exclude the orange population from your gate, but you would end up still sorting both as it will sort anything below that minimum threshold value regardless of if it is gated or not. Again, post sort shows that even though only the green was gated, both green and orange populations are present in the post-sort sample.

We have found this to be true on both the Arias as well as the S6—The S6 allows for up to 16 gating levels vs. the Aria’s limit of 8, but still cannot distinguish gating below 0.
(I set this up with calibrite beads, FITC is green, PE is orange, then unstained and APC are the blue. I purposely overcompensated FITC from APC, and PE from APC, to push the populations below 0 but still be able to distinguish them.)