I have a new-found admiration for bloggers that post daily, or even weekly, as we approach a year since I started drafting this post. So I decided to break it into parts to start telling the story (and saving actual data comparisons for a later date), and hopefully provide some insight to fellow cytometrists.
That which we call a 685LP, by the same name, may not be equivalent!
So I learned last fall (actually now the fall of 2016), when during routine QC on our LSR Fortessa, we were running ultra-rainbow single peak beads and checking that they read 100k, and found that the BV421 channel (only) was down 20k channels. Checking the filters (a multi user core where they are able to swap filters to change the configuation), we found that a different 685 LP filter had been installed on the violet detector array. Simply switching back to the original 685LP brought the signal back to 100k.
At the same time, we were buying some new dichroics for the BUV dyes, and Chroma had two different lots available of a particular wavelength, and sent me the transmission curves, one of which showed significant transmission in the violet region. Clearly I didn’t want THAT one for my application. I did not have a lot number for the questionable filter we had found, but Chroma agreed to scan it for me, and it too showed a violet peak of transmission. Other filters for which I did have a lot number, the vendors have willingly provided the transmission curves for that batch.
The final piece was Paul Robinson posted an article on LinkedIn about this new device, the GoSpectro, which can make your smartphone a spectrometer and was a finalist for the Prism Awards http://www.alphanov.com/223-news-gospectro—the-power-of-spectroscopy-at-your-fingertips.html
There is now a US-based website as well, https://www.gospectro-usa.com/ but I had to wait 3 months for my hospital to approve them as a new vendor out of France. Once I received my GoSpectro, it was quick and easy to look at the transmission of the filters, and even capture images of their transmission properties or use the app to measure it, not as accurately as a dedicated spectrometer but at a fraction of the cost.
When sorrows come, they come not single spies. But in battalions
What I found was that many of our longpass dichroics had this violet transmission, or other colors as well. Here are some example pictures captured with my iPhone using the GoSpectro, not all of them are 685LP, but all of them clearly show undesired (and unexpected!) transmission below the cutoff. The first is the full spectrum that can be visualized, the single violet band is 425/20, and the rest are 635LP or longer, showing a range of transmission below that.
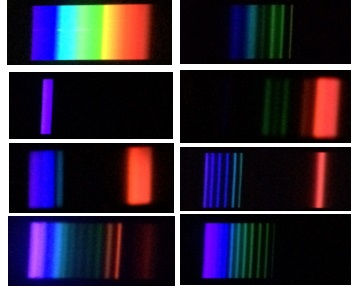
You’ll notice that there is a cutoff on the red side, due to the limitations of the camera not recording infrared light. Some research revealed that newer iPhones have a filter on the camera to block out the IR light, but the iPhone *4* user-facing camera does not! Well, I dug out my old phone, charged it, and working around a cracked screen tried it, but was not able to extend the range of detectable signals with my setup. You can, however, see the IR flash of a remote control, for example. I may have to play with different conditions (different light source, for example–maybe the remote control, itself??) and I’ll post any progress.
I decided to put together a poster about this for CYTO 2017 to share what I had learned, and develop a testing method using the filters on the instrument itself. Placing the bandpass filter for the region you want to be able to measure (so for BV421 you can use the 425/20 bandpass) in front of the first detector, you can then place your different longpass filters in front of it while running multi-peak beads.
What light through yonder filter breaks? It is FITC, but we are measuring PE-Cy5!
If the longpass reflects all of your 425/20 light, then no light will reach the detector and your bright and dim peaks will be a single peak (negative). If there is some transmission of the violet light, then the peaks will resolve themselves. You can get a sense of how much transmission there is by measuring the beads with NO longpass filter, so that all of the 425/20 signal reaches the detector, and setting the voltage so the brightest bead is at 100k (or some other standard). The signal present when a longpass is in place will be some fraction of that total, indicating how much light is getting through.
What this means for your actual data, which is what we should be concerned about, is that as you are losing more and more light to “leaky” filters, your signal at your detector will decrease, and we can look at this impact on the stain index for that marker. Stain index is a calculation of the separation of your peaks, there is a nice writeup of it here or here or here.
This set of plots from the CYTO poster shows a very dim population (0.01 ul of antibody on comp beads, negative and positive collected separately to be able to show the peaks) with all “good” or some “bad” filters in sequence. The “good” filters allow resolution of the populations, but the “bad” filters cannot separate them, even if the voltage is raised to bring the negative population to the same MFI as the “good” filters, with the “good” filters giving 5x the Staining Index of the “bad”.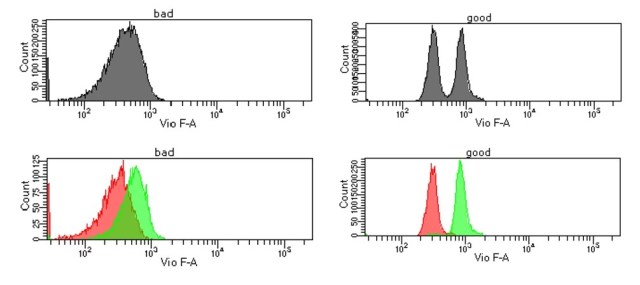
Nothing is good or bad, but thinking makes it so…
In spite of my choice of labels in this post, these filters are not necessarily “bad” or poorly made, they were simply made to certain specifications, such as reflection down to 488, and so may be unsuitable for use on violet or UV excitation lines, while perfectly fine for use on the red, blue or yellow-green lines. Without knowing the specific properties of the filters, it is easy for even an experienced cytometrist to swap in the “same” filter when changing a configuration and end up with different results.
Therein lies the rub.
So now we are left with either labeling all of our filters for their proper locations (“Red laser 685LP”), or replacing all of them with filters that can be used universally. It would be quite an undertaking for a large core to replace ALL of their filters on their instrument so that they are all the same, especially if you have a core like mine where an instrument was added every couple of years, so each one has a different lot of filters on them. I’d love to hear about your instruments and filters in the comments.



