A HUGE Thank you to all of the attendees and vendors that joined us on October 12th for our fall meeting! Presentations are posted to the “Meetings” page, and it looked like everyone had a great time at MEX for the happy hour! The hostess said we were a really fun group and they’d be happy to host us again, so thank you everyone for being on your best behavior!
In over eleventy one months of running our social media accounts, I update half of them half as often as I should like, but I hope you like more than half the posts half as well as they deserve! We will try to post more in the coming year. We were thrilled to have Akos bring his camera, and he was busy all day! Enjoy the pictures on Facebook and Instagram, hopefully more to come. If you have any pictures to share, please send them our way or post them to the page!
Our 2023 fall meeting is only 2 days away! We will be able to provide parking at the 800 Tech Square Garage for $10 for the day, look for the signs for NECyto. Registration and check-in will be in the atrium of 100 Tech Square, vendors please do not enter before 7AM, attendees are invited to come for breakfast starting at 8AM. Talks will begin at 9am in the first floor auditorium of 400 Tech Square across the street. Attendees and vendors are invited to attend a post-meeting gathering at MEX in 500 Tech Square, lets keep the conversations going! We are looking forward to seeing everyone!
Registraion for the 2023 New England Cytometry User group meeting on Thursday, October 12, 2023 is now open! Use the button below to register on Eventbrite:
The meeting will be held at 400 Technology Square in Cambridge MA in the Ragon Institute’s Schwartz Auditorium for a day of talks, and a vendor fair in the atrium of 100/200 Tech Square. Breakfast will be available at 8. Speakers will include Sami Farhi (Broad Institute), Vera Tang (University of Ottawa), David Ng (University of Utah School of Medicine), Peter Lopez (NYU), Thomas Diefenbach (Ragon Institute), and a presentation from the Organizing Committee. Stick around afterwards for the “impromptu” happy hour. The full agenda will be posted to https://newenglandcytometry.com/meetings/ soon!
If you are retired and would like to attend, please contact us directly! Any vendors that are interested in additional sponsorship opportunities, also please reach out. Additional vendor information will be sent to registrants (venue access, shipping information).
Please save the date for the 2023 Fall NECyto meeting, to be held on Thursday October 12, 2023 returning to the Ragon Institute at 400 Tech Square in Cambridge! We will be opening up the registration site in the next couple of weeks, along with announcing the speaker list, but I wanted to assure our community that the meeting is happening and we are looking forward to welcoming everyone to Tech Square one last time, before the big move across the street! Be sure to pick up this year’s t-shirt, having done the visible spectrum:
this year we’re going UV!
We hope you’ll be visible at the meeting!
Mike Waring President, NECyto User Group Director, Ragon Institute Imaging Core Facility
When setting your sort gates in Diva, there are some limitations that users need to be aware of that are not obvious, especially when it comes to gating on populations close to or below the axis.
I first encountered this issue when samples were either overcompensated, or (as discussed in the previous post) when small fluorescent particles pushed the baseline restore artificially negative, so that the population of interest was at or below 0. There is a minimum “upper limit” for your sorting gate, and if you set the region boundary below this, no events will be sorted (your sort rate and collected events will be 0, even though you see events in the gate on the plots). This is true for ANY gate in the hierarchy, so if your “sort gate” is a descendant of a gate that is set too low, you will not register any sorted events.
Setting the gate any lower than shown for APC will result in no events being sorted.
The next limitation to highlight is that Diva cannot distinguish a LOWER boundary that is less than this cutoff—if you are sorting below this minimum limit, then ANYTHING below that is considered “in the gate” regardless of what lower boundary you set. In this example, region P5 is set around only the orange population, but in the post sort analysis on the right, you can see the green population was ALSO sorted.
Lastly, if you were going to try to sort just the green population, you could set at least one point of the gate above that “minimum threshold” value to get around that “minimum threshold” and get events to sort, and exclude the orange population from your gate, but you would end up still sorting both as it will sort anything below that minimum threshold value regardless of if it is gated or not. Again, post sort shows that even though only the green was gated, both green and orange populations are present in the post-sort sample.
We have found this to be true on both the Arias as well as the S6—The S6 allows for up to 16 gating levels vs. the Aria’s limit of 8, but still cannot distinguish gating below 0.
(I set this up with calibrite beads, FITC is green, PE is orange, then unstained and APC are the blue. I purposely overcompensated FITC from APC, and PE from APC, to push the populations below 0 but still be able to distinguish them.)
In the course of running experiments in Diva, you may have found that your negative population ends up well below 0. This came up in a Purdue post in 2009 and Mario Roederer gave an in-depth explanation (https://lists.purdue.edu/pipermail/cytometry/2009-June/037462.html). I’ve put together a write-up with pictures that I share with my users and thought it would be helpful to post it here.
Baseline Correction and potential issues
In order to remove background signals due to unbound fluorophores, dark current, and ambient light, the Diva Software averages signals between events and subtracts this from the total signal. If there is no signal for that parameter, this can result in a negative value for that event.
Most of the time, signals are corrected appropriately and this results in a population mean that is consistent regardless of flow rate. (You may possibly see a higher CV/more spread at higher flow rate due to greater measurement error). Population labels to the right refer to the flow rate used for acquisition of the events in that gate.
Where it can go wrong
Usually threshold is set on FSC– in cases where particles are very different in size, one may be below threshold while the other is on scale (such as cells vs. beads or debris). Because it is below threshold and is not an “event”, the fluorescent signal of the small particle is averaged into the baseline and subtracted from the signals. In this example, negative cells and fluorescent beads are in the same sample. The beads are so small they fall below FSC threshold and are ignored. Their bright fluorescent signal is averaged into the “baseline” which is subtracted from the signal measurement This can lead to your negative populations shifting artificially far into negative values and can lead to problems with your data files. Here the positive APC peak is either a bead that managed to generate FSC above threshold, or stuck to a cell.
Confirming issue:
One way this issue can be illustrated is to run at different flow rates. The higher the flow rate, the more “undetected” particles that will be present, and the higher the baseline error so the further into the negative your population will go. (If your population doesnt go further into the negative with higher flow rates, then it may be a compensation issue).
Solution:
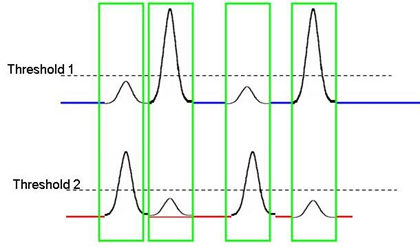
One way to accommodate this is to apply a second threshold using the fluorescent parameter where the bright small particles are occurring (we often see it with PE antibodies that have aggregated):
Applying a second threshold that identifies the small particle based on fluorescence makes sure all events are detected and the baseline is not erroneously high—events above T1 OR T2 will be recognized and recorded. The large APC+ peak below consists of the beads being detected by fluorescence threshold:
Alternatively, you can decrease your FSC threshold to include the debris, but you may not be able to set it low enough to detect all of them, or may end up in the FSC noise. This also defeats the purpose of a higher threshold being used to crop out unwanted debris events in the data file. Another option would be to set a “Save gate” using a FSC threshold, so that the small particles are recognized during acquisition but not saved in the data file.
From BD’s whitepages: 2.5 Baseline Restoration PMT signals can contain a high level of background from a variety of sources: light from unbound fluorophores, PMT dark current, and ambient light. Background signal is eliminated in two stages. The first, gross adjustment is made during the initial conversion of the signal from current to pulse. (Photomultiplier tubes are a source of current.) After pulse conversion in the preamp, the output signal falls in the range of 0 to 5 V. This initial gross adjustment preserves the dynamic range of the A/D converters for signals of interest, and maintains a safety margin of 100 mV. The second, final adjustment is made just before area and height are measured. The FPGAs (Figure 2) calculate a running average of the data outside any window gate to remove the last 100 mV (Figure 3).
Hello NECyto community! October 27 is fast approaching, I hope everyone is as excited as we are. This year’s meeting will return to the Ragon Institute Schwartz Auditorium at 400 Technology Square, with a vendor fair in the atrium of Buildings 100/200 just across the street. Register using this link: https://www.eventbrite.com/e/422824779297
As usual, we will start with breakfast at 8, with talks starting at 9, and then lunch and an afternoon break. We are pleased to offer a stellar lineup of speakers this year, and are planning something a little different for the meeting, but something we should all be used to at this point–we have decided to feature a remote presenter.
Titles to come, but our speakers and topics will be: Jonathan Herman, Ragon Institute: Systems serology in Malaria Virginia Camacho, Boston Childrens Hospital: Tregs in myeloid leukemia Fabienne Lucas and Drew Williamson, BWH: Machine learning in pathology Charlotte Graham, MGH: Clinical CAR-T cell assays Diether Recktenwald, : History of Flow
Our final presentation slot will be dedicated to an open forum for the community to share their ideas on what the NECyto user group can do to best serve the community. We will be asking for members of the community to help out, whether its organizing/posting the monthly happy hours, or finding host sites for periodic seminars. If you’d like to complete our survey, the link is: https://docs.google.com/forms/d/e/1FAIpQLSe9zeg1gZqXpHOYrZi_Z56uoYM259fBxvTWUfI97IVOA_goHA/viewform?usp=sf_link
We will also remember Howard Shapiro, whose absence from the meeting will be sorely felt, and his distinctive closing remarks will be missed. But as he would insist, the FLOW must go on, so we will take the lessons he taught us to do bigger and better science!
We hope you can join us on October 27 for the fall meeting!! Please register using the link below so we know how many to expect. We’ve switched to a new registration site which does add a small convenience charge, but we’re keeping the rate the same as before–$50 for attendee, $600 for vendor. *Vendor table includes 2 attendees, additional representatives are asked to register as a regular attendee.* https://www.eventbrite.com/e/422824779297
Since we are technically a hospital space, we do have some COVID precautions that we will have to follow that I will post to newenglandcytometry.com in the near future. It will include an attestation to vaccination status (COVID and Flu) and any current symptoms. While I’m here I’ll also include the survey form link, please fill it out if you get a chance.
The New England Cytometry (NECyto) organizing committee is currently working on our annual meeting (27 October at the Ragon Institute). This will be our first in-person meeting since 2019, and we are excited to host a collaborative day of cutting-edge cytometry and networking with colleagues.
New England Cytometry was founded to gather scientists, physicians, and shared resource lab members who use these technologies. As our technology and community has grown and changed over the past 27 years, our past leaders have been instrumental to the continuity of this meeting. Without their work, NECyto would not be here today.
We need your help to create a roadmap for the future of NECyto. On Monday we will send a link to a Google Form where you may anonymously submit thoughts and ideas. We will then host an open discussion at this year’s annual meeting. We welcome feedback from all cytometrists – academic researchers, core managers and technicians, biotech scientists – anyone who benefits from our local community.
Hello NECyto members and flow community! I would like to announce that Michele Griffin, who works with Glenn at the Koch Institute Flow Core at MIT, is interested in starting up a new meeting group for interested flow cytometrists.
flowomen will be an informal, fun women’s flow cytometry gathering where members meet up to vent all things flow and support each other as strong women leaders and scientists. Location TBD (a local Cambridge Restaurant), on Wednesday September 7 at 5:30 PM. RSVP to Michele (michelep@mit.edu).
I’ll also take this opportunity to remind you all that BBGs are coming back, we will skip July but the first Monday in August is August 1, so block off your sort calendars now so you can come! As always, you can send me suggestions for venue, and let me know you are coming so I can book a table.
While you have your calendar out, pencil in NECyto for October 27, for the return of our annual fall meeting. Looking forward to seeing you all there!