
So the earlier posts were a lead-in to this one, which I hope will clarify this important issue in compensation. When calculating compensation, it is important to make sure that your single stained control is as bright or brighter than your experimental sample for each color. To illustrate why this is important, a GFP+ sample was measured in 6 detectors off of the blue laser: 525/50 to measure the GFP, and then also in 576/26 (PE), 610/20 (PE-TR), 675/20 (PE-Cy5), 695/40 (PE-Cy5.5), and 780/60 (PE-Cy7) to examine the spillover of GFP into these other detectors. Populations are identified based on their GFP signal (neg, dim, mid, and bright), and plot titles refer to what was used to determine compensation from each channel (none=no compensation, then dim, mid, or high/bright indicates which population was compared to neg to calculate spillover).
Compensation is basically determining the ratio of the two detectors in question: the signal in the primary detector, and then how much signal is detected in another channel (the spillover channel) for that intensity of staining. This allows the researcher to subtract the spillover signal from other channels so that signal from that detector corresponds to that additional parameter. In essence, the slope of the population’s signals is what is being determined:
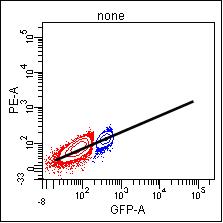
If you are using a dim population, noise in the detection system reduces your sensitivity and so the signal will not be accurately measured. When you look at a brighter population, it will probably not be compensated accurately.
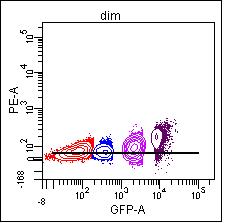
Compensating with the dim population, the resulting means are neg 57, dim 57, mid 89, and bright 182.
In the case of a high spillover between channels, such as GFP spilling into the PE channel, it is possible to get decent compensation of the bright population using the “mid” population, since the signal is high enough to give you a more accurate measurement:
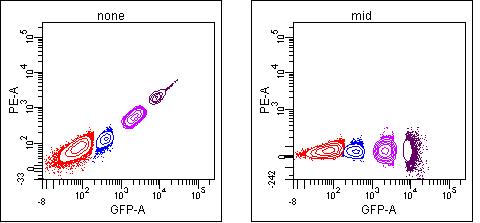
mean: 56, 52, 57, 49
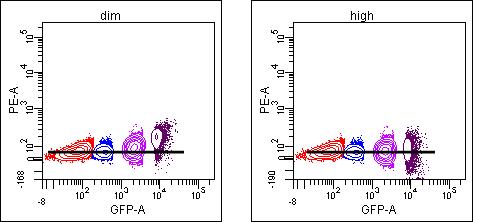
But this may not be true for other spillovers in the same panel. As you move further away from the optimal detector for that fluorochrome, only the brightest events will show spillover that can be detected above background:
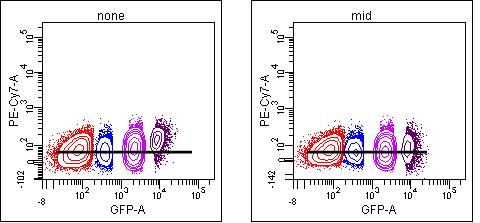
In this case, using the “mid” population results in slight undercompensation of the brights (means of 72, 72, 73, 89) even though it “looks” ok. (Of course, you should never use visualization to set compensation, but compare the means or medians of the negative and positive populations).
Even if different spillover values are generated using bright vs dim populations, the bright values will still work for the dim. Changes in the spillover values have a much greater impact on events with high signal than dim. For comparison, a 1% change in compensation in this example changes the mean signal of the dim by 5, mid by 24, or bright by 102 units in the spillover channel:
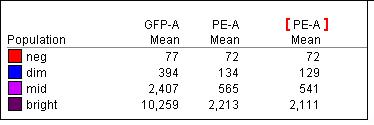
Correcting the spillover from 19.8% using the dim, which leads to the bright being undercompensated by 125 (73% off), to 21.02% using the bright makes the dim “overcompensated” by 4 which is hardly noticeable (5.5% off).
And just for completeness, below are tables of the spillover % for the different setups: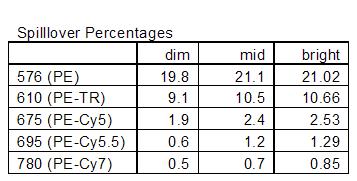
…And the statistics tables for the means (none, dim, mid, bright)





